Sequence Assistant ver.2.x ����}�j���A��
�͂��߂�
Sequence Assistant
�̓���
�̌��ł̋@�\����
���C�Z���X�w�����@
�C���X�g�[�����@�ƃA���C���X�g�[�����@
�����
�ȒP�Ȏg��������
�t�@�C���̓ǂݍ���
����o���s��
�e���͂��s��
�e��@�\�̐���
�����y�f�n�}�̍쐬
����̓ǂݏグ
�k�d�L���\�A�R�h���\�̕\��
������
�Z���N�^�[
�M���b�v���l�����������z��
�����������ʼn������Ă��邩�H
�v���C�}�[�ɓK�����z��̌���
GC���@�̃p�����[�^�[�̕ύX
Tm
�v�Z���@�̑I��
Complementarity
Score �Ƃ�
�v���C�}�[�̕]��
1 OD260 ������̎��ʂ̎Z�o���@
�v���C�}�[�̕��q�ʂ����߂�
�\���t�H�[�}�b�g�̒���
�����y�f�f�[�^�̕ҏW
�ǂݏグ�����̕ύX
���̑�
�o�[�W�����A�b�v����
�ӎ�
��ҘA����
Sequence Assistant �̓���
Sequence Assistant �͎�Ɉ�`�q�g�݊��������ɕK�v�\���ȋ@�\�荞�݂V���v���ł��邱�Ƃ�ڎw������`�q��̓\�t�g�ŁA�����_�Ŏ��̂悤�Ȃ��Ƃ��ł��܂��B
�E����z��̃A�~�m�_�z��ւ̕ϊ�
�E���⍽�z��E�t�z��ւ̕ϊ�
�E�����y�f�n�}�̍쐬
�E�v���C�}�[�ɓK�����z��̌���
�E�v���C�}�[�z��̕]��
�E�M���b�v���l�����������z��
�E����z��̓ǂݏグ
��
�̌��ł̋@�\����
Sequence Assistant �̓V�F�A�E�F�A�ł��B���o�^��Ԃł͑̌��łƂ��ē��삵�܂��B
�̌��łł͓������ꂽ �sample.txt� �Ƃ����t�@�C�������ǂݍ��߂܂���B�܂��A�R�s�[�A�J�b�g�͂ł��܂����A�Z�[�u�A�y�[�X�g���ł��܂���B���̑��̉�͋@�\�͊��S�łƑS�������ł��B
��
���C�Z���X�w�����@
�@�\��������������ɂ̓��C�Z���X�L�[���K�v�ł��B���C�Z���X�L�[�͊�����Ѓx�N�^�[�̃V�F�A���W�T�[�r�X��ʂ��čw�����܂��B
�n�[�h�E�G�A�L�[�ׂ�
�܂��A���̑���ɂ�� Seqence Assistant ���g�p����p�\�R���̃n�[�h�E�G�A�L�[�ׂ܂��B���j���[�� [Help]-[Registration]
��I�����܂��B
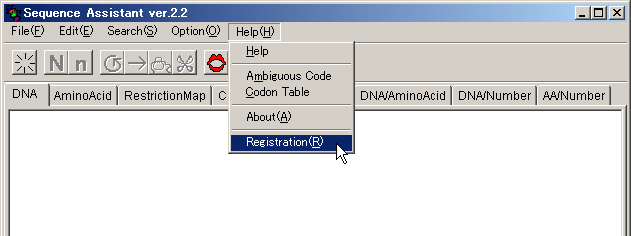
����Ǝ��̂悤�ȃE�B���h�E������܂��B
��i�Ƀn�[�h�E�G�A�L�[���\������܂��̂ŋL�^���Ă����܂��B
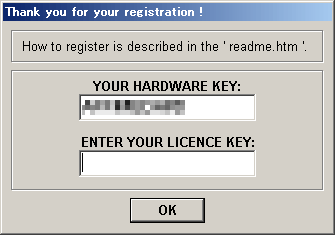
������Ѓx�N�^�[�ōw������
Sequence Assistant �̃��W��i�ԍ��� SR037984 �ł��B
Sequence Assistant �w���y�[�W�ւ̃����N�͂�����ł��B
https://sw.vector.co.jp/swreg/step1.reserve?srno=SR037984
���ׂ��n�[�h�E�G�A�L�[����L�����N��
�u���C�Z���X�L�[�쐬�ɕK�v�ȏ��v �̂Ƃ���ɓ��͂��܂��B
���̑��K�v�ȏ����L�����čw����\�����݁A������Ѓx�N�^�[�̈ē��ɏ]���Ď葱����i�߂�ƁA���͂����n�[�h�E�G�A�L�[�ɑΉ��������C�Z���X�L�[�������Ă��܂��B
���C�Z���X�L�[����͂���
���C�Z���X�L�[���擾������A�Ăя�L�E�B���h�E���J���A���C�Z���X�L�[�����i�ɓ��͂��A[OK]
�{�^���������܂��B���������C�Z���X�L�[�����͂����Ǝ��̂悤�ȃ��b�Z�[�W������܂��B
����œo�^�����ł��B����ȍ~�� Sequence Assistant �̂��ׂĂ̋@�\�����g�������܂��B
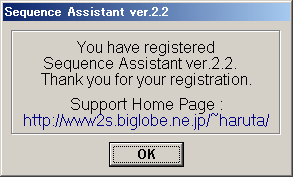
�n�[�h�E�G�A�L�[�͊e�p�\�R���ɌŗL�̒l�ł��B���C�Z���X�L�[�̓n�[�h�E�G�A�L�[�����ɐ�������܂��B
���C�Z���X�̓v���O�������C���X�g�[������p�\�R���̑䐔���w������K�v������܂��B
�n�[�h�E�G�A�\�����ς��ƃn�[�h�E�G�A�L�[���ς�邱�Ƃ�����܂��B���̏ꍇ�͍�҂܂ł����k���������B
��
�C���X�g�[�����@�ƃA���C���X�g�[�����@
�C���X�g�[�����@
���J����Ă��� �seq2x.exe� ���_�E�����[�h���Ď��s����ƃC���X�g�[�����������܂��B
�A���C���X�g�[�����@
�A���C���X�g�[���� Windows �� ��R���g���[���p�l��� �̒��� ��v���O�����̒lj��ƍ폜� ����s���ĉ������B
��
�����
WindowsXP �œ���m�F���Ă��܂��B
��
�ȒP�Ȏg��������
�t�@�C���̓ǂݍ���
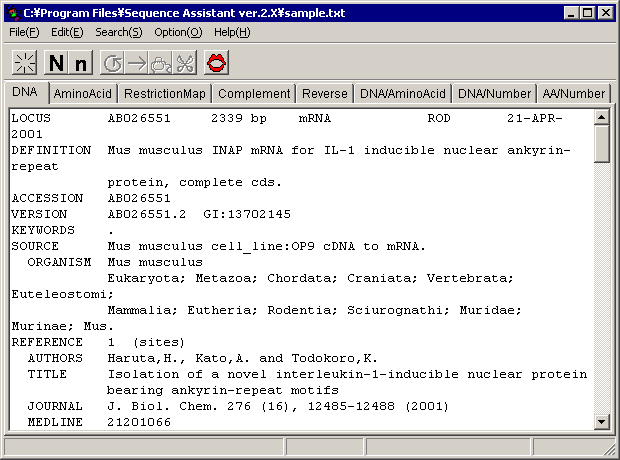
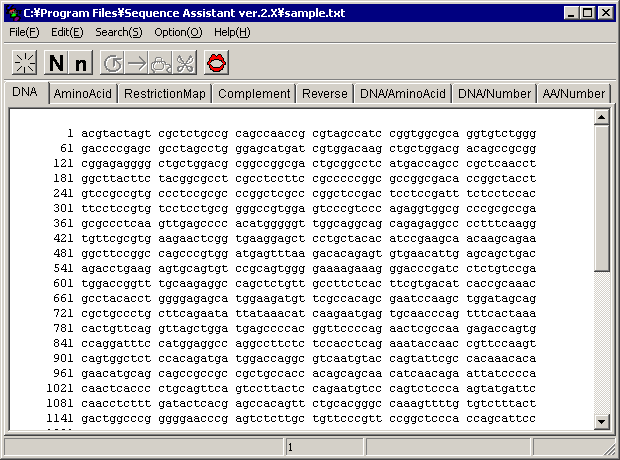
�܂��A�v���O�������N�����ăt�@�C����ǂݍ��ނƎ��̂悤�ɂȂ�܂��B
����o���s��
�Â��āA����z��ȊO�̕������������܂��B
�Â��āA����o�{�^�� �܂���
�܂��� �������܂��B���̑���ł͒P����A,T,G,C ����яk�d�L���� N,R,Y,M,K,S,W,H,B,V,D �݂̂��c���Ă���ȊO�̕������������܂��B�����ɉ���̒����Ƒg����\�����܂��B
�������܂��B���̑���ł͒P����A,T,G,C ����яk�d�L���� N,R,Y,M,K,S,W,H,B,V,D �݂̂��c���Ă���ȊO�̕������������܂��B�����ɉ���̒����Ƒg����\�����܂��B
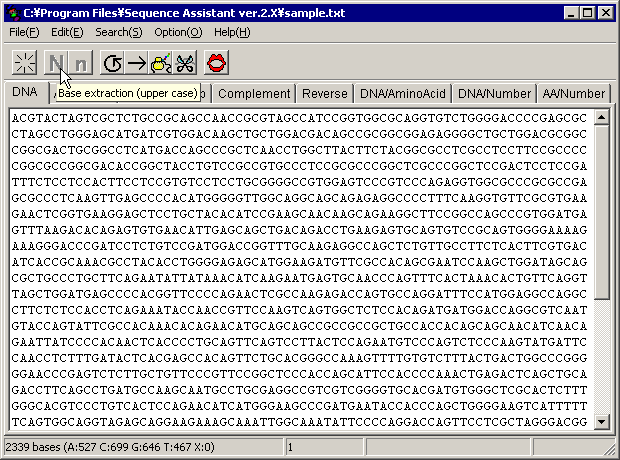
�e���͂��s��
���̌�́A�ړI�ɉ����Ċe�푀����s���܂��B
���̑���̓{�^���������Ǝ��s�ł��܂��B
 : ���Z�b�g�{�^���ł��B�v���O�����̋N������̏�Ԃɖ߂�܂��B : ���Z�b�g�{�^���ł��B�v���O�����̋N������̏�Ԃɖ߂�܂��B
: ����o�{�^���ł��B���̃{�^���͑啶���A�E�͏������ɕϊ����܂��B
 : ���⍽�ɕϊ����܂� (ATGC �� GCAT)�B : ���⍽�ɕϊ����܂� (ATGC �� GCAT)�B
 : �t�z��ɕϊ����܂� (ATGC �� CGTA)�B : �t�z��ɕϊ����܂� (ATGC �� CGTA)�B
 : �A�~�m�_�z��ɕϊ����܂��B : �A�~�m�_�z��ɕϊ����܂��B
 : �����y�f�n�}���쐬���܂��B : �����y�f�n�}���쐬���܂��B
 : ����ǂݏグ���s���܂��B : ����ǂݏグ���s���܂��B
�����̂ق��ɁA���j���[�̒�������s�ł���@�\������܂��B
[Search]-[Primer Search] : �v���C�}�[�ɓK�����z����������܂��B
[Search]-[Primer Check] : �v���C�}�[�z��̕]�����s���܂��B
[Search]-[Similarity Search with Gap] : �M���b�v���l�����������z�����s���܂��B
[Search]-[Search] : ���������s���܂��B�k�d�L���ɂ��Ή����Ă��܂��B
[Edit]-[Selector] :
�w��͈͂̉����I����Ԃɂ��܂��B
|
��
�����y�f�n�}�̍쐬
����o�������
�{�^���������Ɛ����y�f�n�}���쐬����܂��B
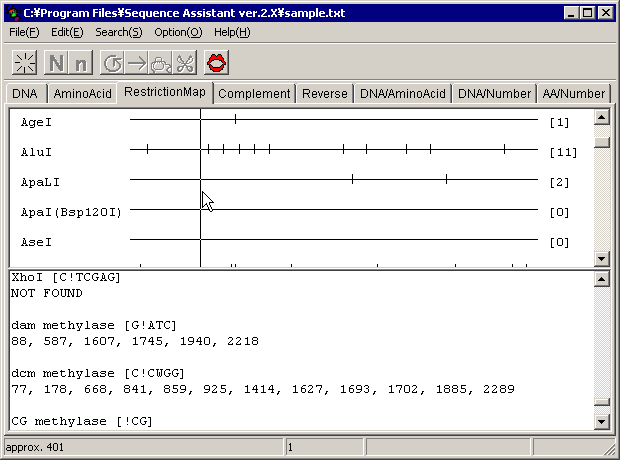
��ʏ㔼���Ő����y�f�n�}���O���t�B�J���\�����܂��B�E�[�́m�n���̐����͐����y�f�̔F�����ʂ̐��ł��B�}�E�X�ƂƂ��ɏc���������܂��B�c���������Ă��邨�悻�̈ʒu���E�B���h�E�����ɕ\������܂��B
��ʉ������Ő����y�f�n�}���e�L�X�g�\�����܂��B�f�t�H���g�ł̓��X�g�̍Ō��dam methylase�Adcm methylase�ACG methylase �ɂ�郁�`�����z����ڂ��Ă��܂��B�Q�l�ɂ��ĉ������BSequence Assistant �̐����y�f�n�}�́A�P�ɔF���z��̈ʒu��\�����Ă��邾���ł��B���`�����̉e���̓��[�U�[�l�ł��m�F�������B
��
����̓ǂݏグ
����̓ǂݏグ�{�^�� �������Ǝ��̂悤�ȓǂݏグ�E�B���h�E���o�Ă��܂��B
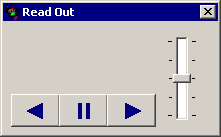
�J�[�\���ʒu���牖��z��������œǂݏグ�܂��B�E���������ɓǂݏグ�A�������ǂݏグ��~�A�����t�����ɓǂݏグ�̃{�^���ł��B�E�̃X�C�b�`���X���C�h������Ɠǂݏグ�X�s�[�h��ύX�ł��܂��B
�ǂݏグ�����͕ύX���邱�Ƃ��ł��܂��B
��
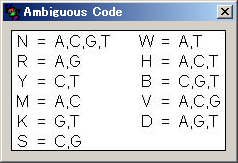
�k�d�L���\�A�R�h���\�̕\��
���j���[�� [Help] �̒��� [Ambiguous Code] ����� [Codon Table] ��I������ƁA���ꂼ��k�d�L���\�A�R�h���\���\������܂��B

��
������
�k�d�L���ɂ��Ή����Ă��܂��B
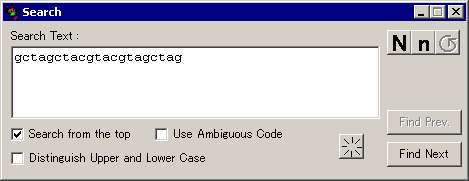
���j���[�� [Search] �̒��� [Search] ��I������Ə�}�̂悤�ȃE�B���h�E���o�Ă��܂��B�����ŕ��������s�����Ƃ��o���܂��B
��
�Z���N�^�[
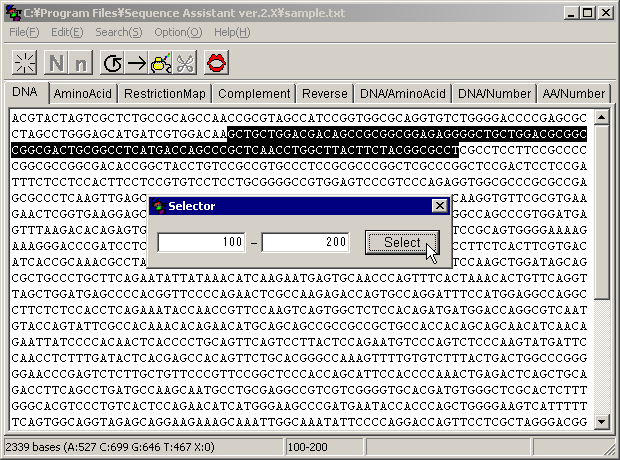
���j���[�� [Edit] �̒��� [Selector] ��I������Ə�}�����̂悤�ȃE�B���h�E���o�Ă��܂��B���ꂪ�Z���N�^�[�ł��B�w�肵���͈͂�I�����邱�Ƃ��ł��܂��B
��
�M���b�v���l�����������z��
�����������ł͏k�d�L���͍l������܂���B
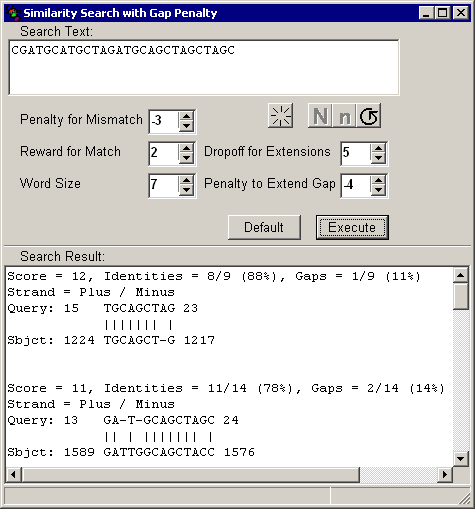
���j���[�� [Search] �̒��� [Similarity Search with Gap] ��I������Ə�}�̂悤�ȃE�B���h�E���o�Ă��܂��B�����ŃM���b�v���l�����������z�����s�����Ƃ��ł��܂��BQuery �z�������o��������Ǝ��s�ł���悤�ɂȂ�܂��B
Word Size ������������ƁA���X�R�A���Ⴂ�z����q�b�g���������ʂ̐��������܂��BWord Size �� Default �� 11 �ł����A�q�b�g�� 0 �������ꍇ�͎����I�� 1 ���Ⴂ�l�ɐݒ肵�Ȃ����čČ������A�������ʂ� 1 �ȏ�ɂȂ����Ƃ���Ō��ʂ�\�����܂��BWord Size �� 1 �ɂ��Ă��q�b�g���o�Ȃ������ꍇ�� Not Found �ƕ\������܂��B
��
�����������ʼn������Ă��邩�H
�����������ł͏k�d�L���͍l������܂���B
�����������ł́A�ȉ��̃p�����[�^���g�p���Ĕ��f���܂��B
�EPenalty for Mismatch (default: -3)
�EReward for Match (default: 2)
�EWord Size (default: 11)
�EDropoff for Extensions (default: 5)
�EPenalty to Extend Gap (default: -4)
�܂��AQuery �z��̐擪���� Word Size �̕�������o���܂��B�����āASubject �z�ɂ��̕����Ȃ����������܂��B
������������������A���̒i�K�̃X�R�A�� [��v����������̒��� (= Word Size)] x [Reward for Match] �̒l�ƂȂ�܂��B�܂��A�����ł͂��̃X�R�A�� [�X�R�A0] �ƌĂԂ��Ƃɂ��܂��B������ Query �z���ł��̕������� 1 ���� 3' ���ɂ����镶�����ASubject �z�Ɍ��������������� 1 ���� 3' ���̕����Ɣ�r���܂��B
�����̕������m����v�����炱�̒i�K�̃X�R�A��
[Reward for Match] �����Z���܂��B��v���Ȃ������ꍇ�́A�@ �~�X�}�b�`�Ƃ��ď��������̒i�K�̃X�R�A�� [Penalty for Mismatch] �����Z����A�A �M���b�v���`�����Ă��̒i�K�̃X�R�A�� [Penalty to Extend Gap] �����Z����A�̗������������܂��B
�����āA����� 1 ���� 3' ���̕������m���r���A[Reward for Match] �� [Penalty for Mismatch] �� [Penalty to Extend Gap] �̂����ꂩ�����Z���܂��B�ȏ�̑�����X�R�A�� [�X�R�A0] - [Dropoff for Extensions] �����������Ȃ�܂ŌJ��Ԃ��܂��B
�~�X�}�b�`�ɂ��邩�A�M���b�v���`�����邩�́A�ŏI�I�ȃX�R�A�������Ȃ�ق����̗p���܂��B
������ Query �z��̐擪���� 1 �������炵�� Word Size �̕�������o���A���l�̑�����J��Ԃ��܂��B����ȍ~�� 3' ���������łȂ��A5' ��������r���Ă����܂��B
�ȏ�̑���� sense ������ anti-sense �����ɂ��Ē��ׂ܂��B
����p�����[�^������ƁA���ړI�ɍ������������������s�����Ƃ��ł��܂��B
�������ʂ̓X�R�A���������ɕ\������܂��B
��
�v���C�}�[�ɓK�����z��̌���
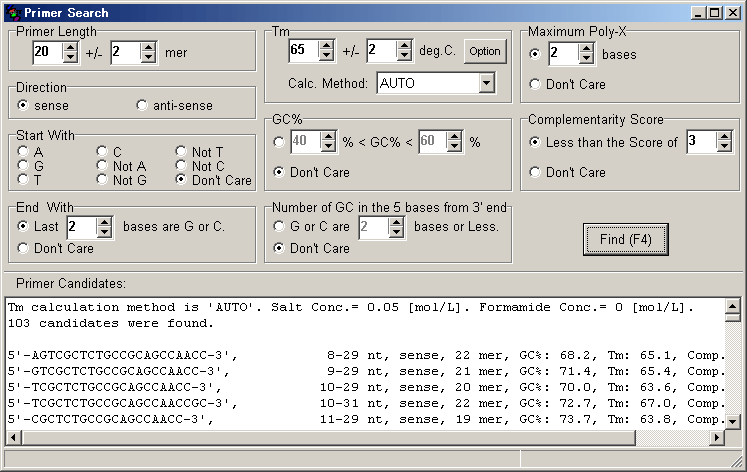
���j���[�� [Search] �̒��� [Primer Search]
��I������Ə�}�̂悤�ȃE�B���h�E���o�Ă��܂��B�����Ńv���C�}�[�ɓK�����z����������邱�Ƃ��ł��܂��B
Maximum Poly-X:
�P�ꉖ���̍ő�A���J��Ԃ������w��ł��܂��B
Complementarity Score (Comp.Score):
Comp.Score �� 0 �ȏ�̒l�����܂����A���̒l���Ⴏ��ΒႢ�قǁA���ȃA�j�[�����O��v���C�}�[�_�C�}�[�̌`�����s�Ȃ��ɂ����Ɨ\�z����܂��B�ʏ�� PCR �ł���� 3
�ȉ��Ȃ�܂���肠��܂���B��҂̓v���C�}�[�_�C�}�[���`�����Ă��܂��v���C�}�[���������Ă��܂������Ƃ�����܂����A���̃v���C�}�[�� Comp.Score ��
10.0 �ł����B
Number of GC in the 5 bases from 3'end:
3' ���[�� 5 ����ɑ��݂��� G �܂��� C �̉�����w��ł��܂��B
��
GC���@�̃p�����[�^�[�̕ύX
���̂����ꂩ�̑�����s���Ɖ��}�̂悤�ȃE�B���h�E���o�Ă��܂��B
�E���j���[�� [Option] - [Tm] ��I������B
�E���j���[�� [Search] - [Primer Search] ��I�����ATm �ݒ�̕����ɂ��� [Option] �{�^���������B
�E���j���[�� [Search] - [Primer Check] ��I�����A[Tm Calc. Method] �ݒ�̉E�ɂ��� [Option] �{�^���������B
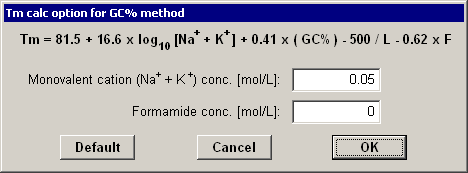
������ Tm �v�Z���@�̂ЂƂł��� GC% �@�̃p�����[�^�[��ύX�ł��܂��B�悭�킩��Ȃ��ꍇ�͏����l�̂܂܂ɂ��Ă����܂��傤�B���}�ɂ����āA������ [Default] �{�^���������A����ɉE���� [OK] �{�^���������Ə����l�ɖ߂�܂��B��ʓI�� PCR �o�b�t�@�[�̉��Z�x�� 0.05 [mol/L] �ł���A�z�����A�~�h�͊܂܂�Ă��Ȃ��̂ŁA���ꂼ��̏����l�� 0.05 [mol/L]�A0 [mol/L] �ƂȂ��Ă��܂��B
��
Tm �v�Z���@�̑I��
�ȉ��̂����ꂩ�̑�����s���Ɖ��}�̂悤�ȃ{�b�N�X���o�Ă��܂��B
�E���j���[�� [Search] - [Primer Search] ��I������BTm �ݒ�̕����ɉ��}�̂悤�� [Tm Calc. Method] ��ݒ肷��{�b�N�X������܂��B
�E���j���[�� [Search] - [Primer Check] ��I������Əo������E�B���h�E�̒��������ɉ��}�̂悤�� [Tm Calc. Method] ��ݒ肷��{�b�N�X������܂��B
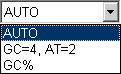
�v���C�}�[�z��̈��: 5'-TTg CgC TgT CTC Tgg Tgg CTC C-3' �̏ꍇ�ɂǂ̂悤�Ȍv�Z���s�����������܂��B
AUTO:
���܂������[�h�ł��B�悭�킩��Ȃ��ꍇ�͂��̃��[�h�ɂ��Ă����܂��傤�B�܂��AGC%�@��Tm�l���v�Z���܂��B�v�Z�̌��ʁATm�l��20���������������炻�̂܂܍̗p���܂��B20�������Ⴉ�����ꍇ�ɂ́AGC=4���AAT=2�� �Ƃ��čČv�Z���A���̒l���g�p���܂��B��ɂ������v���C�}�[�̏ꍇ�� GC% �@�̌��ʂł��� 63.3�� ���̗p���܂��B
GC=
4, AT= 2:
���GC=4���AAT=2�� �Ƃ��������Z�Ōv�Z���܂��B
��ɂ������v���C�}�[�̏ꍇ�A
Tm = 2+2+4+4+4+4+2+4+2+4+2+4+2+4+4+2+4+4+4+2+4+4
= 72��
�ɂȂ�܂��B
GC%:
��� GC% �@�Ōv�Z���܂��B�v�Z���͎��̒ʂ�ł��B
Tm = 81.5 + 16.6 �� log10 ([Na+] + [K+]) + 0.41(GC%) - 500/L-0.62 x F
�����ŁA
[Na+] + [K+] --- Na+ �� K+ �̃����Z�x�̍��v [mol/L]�B
GC% --- G ����� C �̊��� [%]�B
L --- ��{�����`�����镔���̃v���C�}�[�̒����B
F --- �z�����A�~�h�̃����Z�x [mol/L]�B
Tm �I�v�V�����̐ݒ肪�A
�o�b�t�@�[���̈ꉿ�̗z�C�I�� (Na+�AK+) �̔Z�x = 0.05 [mol/L] (�����l)
�o�b�t�@�[���̃z�����A�~�h�Z�x = 0 [mol/L] (�����l)
�ɐݒ肳��Ă���ꍇ�A��ɂ������v���C�}�[�� Tm �l�́A
Tm = 81.5 + 16.6 �� log10 (0.05) + 0.41 x (63.6) - 500/22-0.62 x 0
= 81.5 + 16.6 x (-1.3) + 26.1 - 22.7
= 63.3��
�ɂȂ�܂��B
��
Complementarity Score �Ƃ�
���̋@�\�ł͏k�d�L���͍l������܂���B
Comp.Score
�͓��ꕪ�q���ł̎��ȃA�j�[���̂��₷�� (2 ���\���̎��₷��) ��v���C�}�[�_�C�}�[�̌`���̂��₷���̖ڈ��ƂȂ�X�R�A�� 0
�ȏ�̒l�����܂��B���̒l���������قǁA���ȃA�j�[�����O��v���C�}�[�_�C�}�[�̌`�����s�Ȃ��ɂ����Ɨ\�z����܂��B�ʏ�� PCR ���ړI�ł���� 3
�ȉ��̔z���I������܂���肠��܂���B�v���C�}�[�����@�\��A�v���C�}�[�]���@�\�Ŏg�p���Ă��܂��B
Comp.Score �̎Z�o���@
1
�̉���z��ɂ��ĎZ�o����ꍇ
�Ⴆ�A5'-ATCGGCTACTAGTCAATGTG-3'
�Ƃ�������z��ɂ��Čv�Z����ꍇ�́A���̂悤�Ȃ��Ƃ��s�Ȃ��Ă��܂��B�܂��A���̉���z������̂悤�ɕ��ׂ܂��B
5' ATCGGCTACTAGTCAATGTG 3'
3' GTGTAACTGATCATCGGCTA 5'
�����āA�����珇�Ɍ݂��ɑ���I�ɂȂ鉖��Ɉ�����܂��B
5' ATCGGCTACTAGTCAATGTG 3'
| |||||| |
3' GTGTAACTGATCATCGGCTA 5'
������ Comp.Score
���v�Z���܂��B�����珇�Ɍ��čs���A�݂��ɑ���I�Ȃ� +1�A�����łȂ���� -1 �Ƃ��������Z���s���܂��B���̏ꍇ�͎��̂悤�ɂȂ�܂��B
-1-1+1-1-1-1-1+1+1+1+1+1+1-1-1-1-1+1-1-1 = -4
Comp.Score �͍ŏ��l�� 0 �Ƃ��܂��B�}�C�i�X�̒l���o�Ă� 0 �ł��B�]���āA���̏ꍇ�� Comp.Score �� 0 �ƂȂ�܂��B
���ɁA�㑤�̔z��� 1 ����E�ɂ��炵�ē��l�̌v�Z���s�Ȃ��܂��B
5' ATCGGCTACTAGTCAATGTG 3'
|
|
3' GTGTAACTGATCATCGGCTA 5'
���̏ꍇ�� Comp.Score �͎��̂悤�Ȍv�Z���� 0 �ƂȂ�܂��B
+1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1+1 = -15
�����ď㑤�̔z�������� 1 ����E�ɂ��炵�� Comp.Score ���v�Z���܂��B
5' ATCGGCTACTAGTCAATGTG 3'
| |
3' GTGTAACTGATCATCGGCTA 5'
����𑱂��čs���ƁA�Ō�͂����Ȃ�܂��B
5' ATCGGCTACTAGTCAATGTG 3'
3' GTGTAACTGATCATCGGCTA 5'
�����܂ŗ�����A�܂����[�𑵂��āA���x�͉����̔z��� 1 ����E�ɂ��炵�� Comp.Score ���v�Z���܂��B���̏ꍇ�� Comp.Score �� 0 �ł��B����ɑ����܂��B
5' ATCGGCTACTAGTCAATGTG 3'
| |
3' GTGTAACTGATCATCGGCTA 5'
����𑱂��čs���ƁA�Ō�͂����Ȃ�܂��B
5' ATCGGCTACTAGTCAATGTG 3'
3' GTGTAACTGATCATCGGCTA 5'
���̔z��� Comp.Score
�́A�ȏ�̈�A�̑���̌v�Z���ʂ̂����̍ő�l�ł��B���̏ꍇ�͎��̎��� 7.0 �ƂȂ�܂��B
5' ATCGGCTACTAGTCAATGTG 3'
|| | ||| ||| | ||
3' GTGTAACTGATCATCGGCTA 5'
2 �̉���z��ɂ��ĎZ�o����ꍇ
�v���C�}�[�]���@�\�Ŏg�p���Ă��܂��B���@�͊�{�I�� 1 �̉���z��ɂ��ĎZ�o����ꍇ�Ɠ����ł��B�܂��A2 �̉���z�ꂼ��ɂ��āAComp.Score ���Z�o���܂��B
������ 2 �̉���z�����ׁA1 �̉���z��ɂ��čs�Ȃ����̂Ɠ��l�� 1 ��������炵�Ȃ���AComp.Score ���v�Z���܂��B
�ȏ�̑����S�čs�Ȃ�����A����ꂽ Comp.Score �̍ő�l�� Comp.Score �Ƃ��ĕ\�����܂��B
���̂悤�ɂ��āA2 �̉���z��Ō`�����꓾�邷�ׂĂ̑g�ݍ��킹�ɂ��Čv�Z���s�Ȃ��܂��B
��
�v���C�}�[�̕]��
���̋@�\�ł͏k�d�L���͍l������܂���B
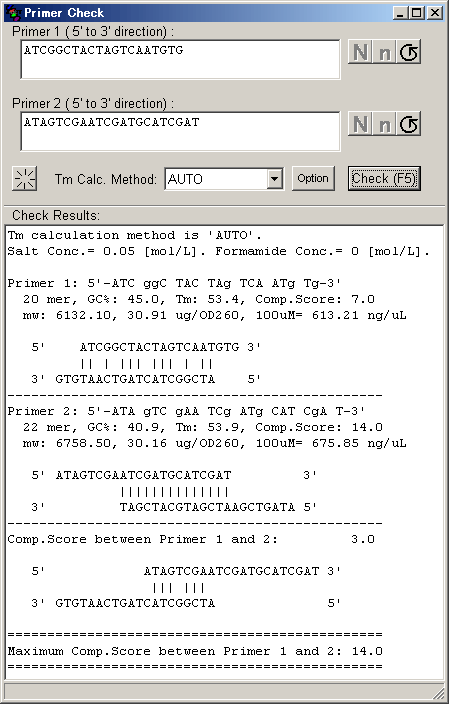
���j���[�� [Search] �̒��� [Primer Check] ��I������Ə�}�̂悤�ȃE�B���h�E���o�Ă��܂��B�����Ńv���C�}�[�z���]�����邱�Ƃ��ł��܂��B�����ł́A2 �̔z���]�������ꍇ�̗�������Ă��܂��B
�܂��A�]�������v���C�}�[�z��� 3 �c��Ƃɋ���ċ���� 'A,T,C' �͑啶���� 'g' �͏������ŕ\�����܂��B�����ăv���C�}�[�̒����AGC�ܗʁAComp.Score�A�v���C�}�[�̕��q�ʁA�v���C�}�[�� 1 OD260 ������̎��� [ug]�A100uM �n�t�ɒ�������ꍇ�̃v���C�}�[�̔Z�x [ng/uL] ��\�����܂��B
������ Comp.Score �̍ő�l�����n�C�u���_�C�Y�̃p�^�[����\�����܂��B��}�� Primer 2 �̓v���C�}�[�_�C�}�[���`�����邱�Ƃ��킩��܂��B
1 OD260 ������̎��ʂ̎Z�o���@
�Ⴆ�A5'-ATCGGCT-3'
�Ƃ�������z��ɂ��Čv�Z����ꍇ�́A���̂悤�Ȃ��Ƃ��s�Ȃ��Ă��܂��B
�܂��A�ȉ��̎��ɂ��v���C�}�[�̕��q�ʂ��v�Z���܂��B
A �c��̐� x 313.2 + C �c��̐� x 289.2 + G �c��̐� x 329.2 + T �c��̐� x 304.2 - 61.9
��̉���z��̕��q�ʂ� 2096.50
�ƂȂ�܂��B
�����āA���̕\����אڂ��鉖��� Pairwise
�z���W�������߁A���v���܂��B����� UV �l�Ƃ��܂��B
Pairwise �z���W��
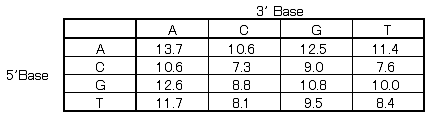
��̉���z��̏ꍇ�A
11.4 +
8.1 + 9.0 + 10.8 + 8.8 + 7.6 = 55.7 (UV �l)
�����āA���̕\���痼�[�̉�����������S�Ẳ���̋z���W�������߁A���v���܂��B����� Eb �l�Ƃ��܂��B
����̋z���W��
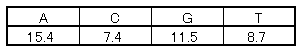
��̉���z��̏ꍇ�A���[�� A ����� T ������ 5 ����̋z���W�������v���܂��B
8.7 + 7.4 + 11.5 + 11.5 + 7.4 = 46.5 (Eb �l)
���̌v�Z���ɏ�L�� UV �l����� Eb �l�Ă͂߂܂��B���̒l�� [ug/OD260] �ɂ�����܂��B
���q�� �� (2 �~ UV �l - Eb �l)
= 2096.50 �� (2 �~ 55.7 - 46.5)
= 32.30 [ug/OD260]
�ƂȂ�܂��B
��
�\���t�H�[�}�b�g�̒���
���j���[�� [option] �̒��� [view] ��I������Ǝ��̃E�B���h�E���o�����܂��B�����ŁADNA/AminoAcid�ADNA/Number�AAminoAcid/Number �̊e�y�[�W�� 1 �s������̕�������ς��邱�Ƃ��ł��܂��B
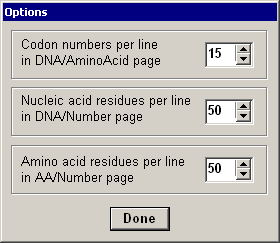
��
�����y�f�f�[�^�̕ҏW
�����y�f�f�[�^�̃t�H�[�}�b�g�ɖ�肪����� Sequence Assistant
���듮�삷��\��������܂��B�ҏW�̓��[�U�[�l�̎��ȐӔC�ōs���Ă��������B
�����y�f�f�[�^��ҏW����O�ɃI���W�i���f�[�^���o�b�N�A�b�v����邱�Ƃ������߂��܂��B
�����y�f�f�[�^�̓e�L�X�g�t�@�C���ŁASequence Assistant �̖{�̂ł���useq_20.exe�v�Ɠ����t�H���_�ɂ���܂��B�t�@�C�����́uredata.dat�v�ł��B
�f�t�H���g�ł̓��X�g�̍Ō�� dam methylase�Adcm methylase�ACG methylase �ɂ�郁�`�����z����ڂ��Ă��܂��B�Q�l�ɂ��ĉ������B
�uredata.dat�v�t�@�C���̃t�H�[�}�b�g�ɂ͎��̂悤�Ȍ��܂肪����܂��B
�E�s���Ɂu//�v��t���邱�Ƃɂ��R�����g�A�E�g�ł��܂��B
�E�s���ʼn��s���Ă���肠��܂���B
�E�u�����y�f��+�R���}+�F���z��+���s�v������ꂸ�ɏ����܂��B�F�����ʂ��u!�v�ŕ\�����Ȃ���Ȃ�܂���B
�E�t�@�C���́useq_20.exe�v�Ɠ����t�H���_�ɑ��݂���K�v������܂��B
�ȉ��ɕW���œY�t����Ă��� �uredata.dat�v�̓��e�������܂��B
------------------------- �������� -------------------------
//
// Restriction Endonucleases data for Sequence Assistant software.
//
AatII,GACGT!C
AccI,GT!MKAC
AccII(FnuDII),CG!CG
AccIII,T!CCGGA
AflII,C!TTAAG
AgeI,A!CCGGT
AluI,AG!CT
ApaLI,G!TGCAC
ApaI(Bsp120I),G!GGCCC
AseI,AT!TAAT
AvaII,G!GWCC
BalI,TGG!CCA
BamHI,G!GATCC
BclI,T!GATCA
BglI,GCCNNNN!NGGC
BglII,A!GATCT
BssHII,G!CGCGC
BstXI,CCANNNNN!NTGG
DraI(AhaIII),TTT!AAA
EcoRI,G!AATTC
EcoRII(MvaI),CC!WGG
EcoRV,GAT!ATC
FspI,TGC!GCA
HaeIII,GG!CC
HhaI,GCG!C
Hin6I,G!CGC
HindIII,A!AGCTT
HinfI,G!ANTC
HpaI,GTT!AAC
KpnI,GGTAC!C
MluI,A!CGCGT
MspI(HpaII),C!CGG
NarI(BbeI),GG!CGCC
NciI,CC!SGG
NcoI,C!CATGG
NdeI,CA!TATG
NdeII(MboI),!GATC
NheI,G!CTAGC
NotI,GC!GGCCGC
NruI,TCG!CGA
NsiI,ATGCA!T
NspV,TT!CGAA
PstI,CTGCA!G
PvuII,CAG!CTG
RsaI(Csp6I),G!TAC
SacI(SstI),GAGCT!C
SacII(SstII),CCGC!GG
SalI,G!TCGAC
Sau3AI,!GATC
ScaI,AGT!ACT
SmaI,CCC!GGG
SpeI,A!CTAGT
SphI,GCATG!C
SspI,AAT!ATT
StuI(AatI),AGG!CCT
StyI,C!CWWGG
TaqI,T!CGA
XbaI,T!CTAGA
XhoI,C!TCGAG
dam methylase,G!ATC
dcm methylase,C!CWGG
CG methylase,!CG
------------------------- �����܂� -------------------------
��
�ǂݏグ�����̕ύX
�ǂݏグ�����̃t�@�C���ɖ�肪����� Sequence Assistant
���듮�삷��\��������܂��B�ύX�̓��[�U�[�l�̎��ȐӔC�ōs���Ă��������B
�ǂݏグ������ύX����O�ɃI���W�i���f�[�^���o�b�N�A�b�v����邱�Ƃ������߂��܂��B
����ǂݏグ�̐��� wav �t�@�C���Ŏ��� 5 ��ނ���Ȃ�ASequence Assistant �̖{�̂ł��� �useq_2x.exe�v �Ɠ����t�H���_�ɂ���܂��B
�Ea.wav : a �ɑΉ����܂��B
�Et.wav : t �ɑΉ����܂��B
�Eg.wav : g �ɑΉ����܂��B
�Ec.wav : c �ɑΉ����܂��B
�Eerrror.wav :
��L�ȊO�ɑΉ����܂��B
�����̃t�@�C�������D���� wav �t�@�C���Œu��������Γǂݏグ������ύX���邱�Ƃ��ł��܂��B�������A���̂悤�Ȍ��܂肪����܂��B
�E�t�@�C������ύX���Ă͂����܂���B
�E5 ��ނ̃t�@�C�������ׂđ����Ă���K�v������܂��B
�E�t�@�C���� �useq_2x.exe�v �Ɠ����t�H���_�ɑ��݂���K�v������܂��B
��
�o�[�W�����A�b�v����
ver.2.1 �� ver.2.2
�E�v���C�}�[�����@�\���������܂����B
�E�E�N���b�N���j���[��t���܂����B
�E�v���C�}�[�̕]����ʂŁA�v���C�}�[�̕��q�ʂ�\������@�\��t���܂����B
�E�v���C�}�[�̕]����ʂŁA1 OD260 ������̎��� [ug/OD] ��\������@�\��t���܂����B
�E�v���C�}�[�̕]����ʂŁA100uM �n�t�ɒ�������ꍇ�̃v���C�}�[�̔Z�x [ng/uL] ��\������@�\��t���܂����B
�E�w���v�� html �`���ɂ��܂����B
ver.2.0 �� ver.2.1
�ETm�̌v�Z���@�����ǂ��܂����B���܂ł�GC=4���AAT=2�� �Ƃ����v�Z�݂̂����ł��܂���ł������AGC% �@�ɂ��v�Z���ł���悤�ɂ��܂����B�v�Z�ɗp����Ώۃo�b�t�@�[���̉��Z�x�̒l���ύX���邱�Ƃ��ł��܂��B
ver.1.5.3 �� ver.2.0
�E�M���b�v���l�����������������@�\��t���܂����B
�E[Selector] �@�\��t���܂����B�w�肵���͈͂̉���z���I����Ԃɂ��邱�Ƃ��ł��܂��B
�E[Primer Check] �ŁA�\�z�����v���C�}�[���m�̃n�C�u���_�C�Y�p�^�[����\������悤�ɂ��܂����B
�E�����y�f�n�}���O���t�B�J���ɕ\������悤�ɂ��܂����B
�E�t�z��ւ̕ϊ��@�\��t���܂����B
�E�ǂݏグ�@�\�œǂݏグ�X�s�[�h�̕ύX���ł���悤�ɂ��܂����B
�E[Search] �@�\�����ǂ��A�O�ア����̕����ɂ������ł���悤�ɂ��܂����B
��
�ӎ�
Seauence Assistant �̃C���X�g�[���v���O�����̍쐬�� nobukichi ���́u�ȒP�C���X�g�[���v�𗘗p�����Ē����܂����B���̏����Ă���\���グ�܂��B
nobukichi ���̃z�[���y�[�W�F
http://www5a.biglobe.ne.jp/~nobukich/
��
��ҘA����
��҂̃��[���A�h���X�� haruta@mx3.mesh.ne.jp
�ł��B
�\�t�g�Ɋւ��邲����₲�v�]�Ȃǂ��C�y�ɂ��������B
��
2006/5/31
2009/5/17
�C��